
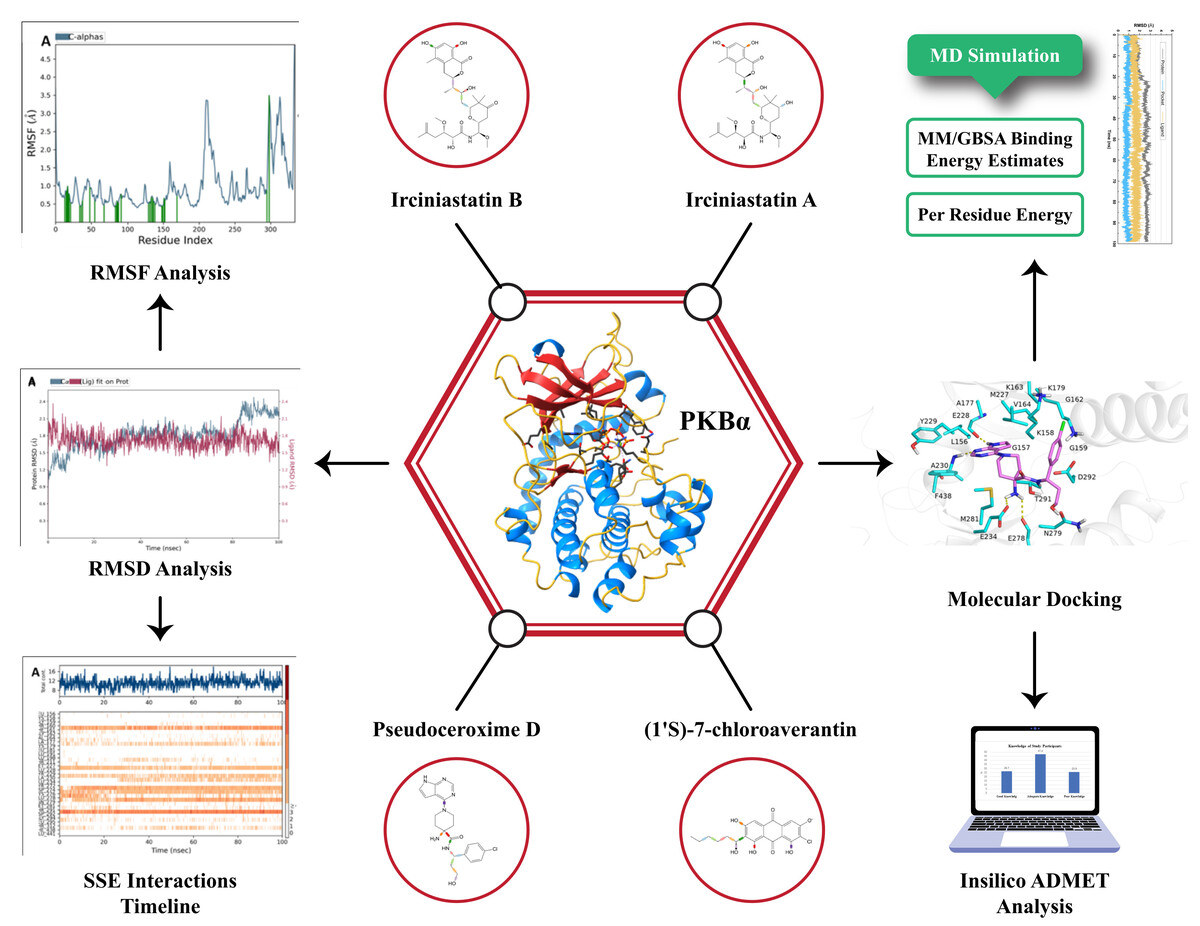

Introduction:
Cancer is a prominent cause of mortality worldwide, ranking second in terms of fatality rates and resulting in around 9.6 million deaths during the last several decades, and is responsible for around one in every six deaths worldwide (Feng, Zong, Cao, & Xu, 2019). According to the World Health Organization (WHO), the number of new cases would rise by nearly 70% over the next two decades, from 14 million to 22 million (Shahzadi et al., 2021), and it was the leading cause of death for 10 million people in 2020 (Sung et al., 2021). At now, cancer treatment options include chemotherapy, surgery, and radiation therapy. Cancer patients have a pressing need and desire for the discovery of innovative compounds that can destroy tumor cells and halt their development without causing severe side effects to the body, as is the case with present chemotherapeutic treatments. As a result, the development of novel cancer treatments is a major focus of the researchers and pharmaceutical industry (Roney et al., 2023).
In recent years, research has concentrated on discovering new chemical entities from natural sources in order to meet the immense need for novel kinase inhibitors while also addressing crucial medical diseases, including cancer with connection to signal transduction pathways (Gill, Saleem, & Ahemad, 2020). One of these protein kinases is the serine/threonine protein kinase, i.e., protein kinase B (PKB, also known as Akt). It is a target of phosphatidylinositol 3-kinase (PI3K) that plays a crucial function in the PI3K-Akt-mTOR pathway (Barnett, Bilodeau, & Lindsley, 2005). Human PKBs are classified into three subtypes: PKB/Akt-1, PKB/Akt-2, and PKB/Akt-3, which share over eighty percent homology (Fayard, Tintignac, Baudry, & Hemmings, 2005; Wang et al., 2008). The main isoform PKBα is expressed at higher levels (Thakur, Kumar, Kumar, & C, 2010) and has been a focus for anti-tumor treatment. Activated PKBα can control cell signalling processes by phosphorylating endogenous substrates which are involved in glucose metabolism, cell growth, survival, apoptosis, cell migration, and transcription (Chen, Cao, Chen, & Chen, 2013). The activity of PKBα is often increased in tumors for many reasons. These include the amplification and functional alterations of receptor tyrosine kinases and PI3K, which are upstream regulators of PKBα. Additionally, the loss of PTEN, a negative regulator of PKBα, can also contribute to its heightened activity in tumors. Anomalous PKBα signaling has been detected in several types of human malignancies, such as breast, prostate, ovarian carcinoma, and melanoma (Fayard et al., 2005). Several PKBα inhibitors have been investigated in clinical trials for the treatment of tumors, including GSK690693 and GDC-0068, which are ATP-competitive inhibitors (Blake et al., 2012), however, the clinical phase I trial of GSK690693 was terminated in 2010 (Chen, Cao et al. 2013). There is still a need to find novel PKBα with an enhanced binding affinity towards PKBα, without causing adverse effects.
Natural products are a valuable and cost-effective resource in the advancement of novel pharmaceuticals, with numerous plant-derived chemicals already being utilized in clinical settings for diverse therapeutic purposes (Ain, Batool, & Choi, 2020). The utilization of natural product-derived chemicals and semi-synthetic compounds is a significant avenue for the discovery of novel drug classes (Sinha, Doble, & Manju, 2018). In recent years, the field of virtual screening is undergoing tremendous advancements and is serving as a valuable supplement to the high-throughput screening platform in the realm of drug discovery. The presence of these computer platforms facilitates the screening of chemical databases in order to uncover potential matches for the specified targets (Wang et al., 2020). Molecular docking is widely employed as a computational technique in current drug development due to its shown efficacy in accurately predicting ligand binding interactions. Molecular docking is a computational technique that enables the determination of the binding affinity between drugs and protein targets, as well as the elucidation of the underlying mechanism governing this interaction (Sinha et al., 2018). The computational technique is widely regarded as a trustworthy methodology for the large-scale screening of many compounds in order to assess their biological activities and elucidate the mechanisms through which ligands interact with protein targets (Waltenberger et al., 2016). The utilization of molecular dynamics (MD) simulations enables a comprehensive examination of the stability and feasibility of binding mechanisms exhibited by the substances. The ADMET characteristics are also computed in order to assess the drug-like qualities of the molecule (Alam et al., 2018). In the present study, we have determined the key structural requirements for protein-ligand interactions of PKBα, for the first time, using an arsenal of computational techniques: ligand (shape)-based virtual screening, molecular docking, molecular electrostatic potential (MESP) analysis, MD simulation, and binding free energy calculation to identify highly potent PKBα inhibitors. Additionally, MD simulation, H-bond occupancy analysis, and binding free energy calculations using implicit MM(GB/PB) SA model were performed for three inhibitors with different potency for PKBα. The results obtained in this study can help to identify potential structural and pharmacophoric features governing the binding process and provide further insights into the key structural modifications for the rational design of novel, potent, and selective PKBα inhibitors.
Experimental Section
Target protein and Retrieval of compound structures
The Crystal structure of PKBΑ alpha in complex with AZD536 (PDB: 4GV1) with a resolution of 1.49 Å (Addie et al., 2013) was retrieved from the protein data bank (PDB) and used for molecular docking studies against the selected compounds from the natural product database. The 3D structures of 1651 compounds were extracted from the Database.
Preparation of target protein
The three-dimensional structures of the target proteins were prepared using Maestro module 13.7 (Maestro version, 2023-3) in the Schrodinger suite 2023-3. The Crystal structure of PKBΑ alpha in complex with AZD536 (PDB: 4GV1) with a resolution of 1.49 Å was prepared using the Protein Preparation Wizard in the Maestro module with the force field of OPLS4. The protein preparation process includes deleting water molecules, adding hydrogen atoms, side chains, and missing loops, and removing unwanted metal atoms/ions. The ionization and tautomeric states of the hetero-groups were adjusted. Finally, the optimization steps included optimizing the hydrogen bonds and refining the structure by limiting the RMSD to 0.3 Å. The active binding site residues of PKBΑ alpha (PDB: 4GV1) were identified using the co-crystal ligand AZD536.
Ligand preparation
Retrieved compounds were prepared using the LigPrep (Schrodinger Release, 2014) module available in the Schrodinger Suite. All the ligands were minimised with the force field of OPLS4. A minimum of 32 poses were generated for each ligand. The prepared ligands were further used for molecular docking studies with the target proteins.
Molecular docking protocol
Molecular docking analysis of the target protein PKBα in complex with AZD536 with compounds (ligands) was performed using the Maestro module in the Schrodinger Suite 2023-3 version. The protein was prepared using Prepwiz, and ligands were prepared using LigPrep in Schrödinger. The grid box was constructed using Receptor Grid generation on the centroid of the protein, and protein-ligand docking was performed using SP (Standard Precision) mode. The top ten percentage of the ligands were further docked using the Extra Precision (XP) mode. The docking output file was analyzed using the Maestro software.
ADME/Tox
The QikProp module of Maestro Version 13.7 is an absorption, distribution, metabolism, and excretion (ADME) prediction tool that can make certain descriptors of ADME. It makes predictions for both pharmacologically and physicochemically significant descriptors. Based on Lipinski's rule of five, ADME qualities evaluate the drug-like action of ligand molecules. Maestro Version 13.7 was used to analyze the ADME/T characteristics of the designed compound (Shaikh & Siu, 2016).
Molecular Mechanics-Generalize Born Surface Area (MM-GBSA)
Molecular docking analysis does not show the relative binding energy or free energy of each ligand or the affinity between the protein and ligand complexes (Pattar, Adhoni, Kamanavalli, & Kumbar, 2020). Therefore, MM-GBSA was used to determine the binding energy and free energy between the molecules. It was calculated using Prime MM-GBSA in the Maestro module by uploading the molecular docking output file (. mae format) as the input file.
Simulation of ligand-receptor interaction
The top-scoring compounds were further evaluated for their stability in ligand-receptor interactions. The top-scoring compounds in the form of protein-ligand complexes were considered. SPC was chosen as the solvent model, and an orthorhombic box was selected. The box size was determined based on the buffer volume, which was minimized before the system was built. The salt concentration of 0.15M with Na+ and Cl- was added to the system, and the force field used was OPLS4. The simulation of the prepared ligand-protein complex in the system was run for 100 ns with an energy level of 1.2, and approximately 1000 frames were generated. The ensemble class used was NPT, and the model was relaxed prior to the simulation. The Desmond module available in Maestro v13.7 (Schrodinger v2023-3) was used in this study.
Results and Discussion
Molecular docking
The co-crystal ligand AZD5363 formed 4 H-bond interactions within the binding pocket. Two of these H-bonds are formed by the pyrazolopyrimidine functionality with GLU228 and ALA230 residues in a donor-acceptor configuration respectively. Both of these amino acids lie within the hinge region, surrounded by a hydrophobic cavity. In addition, this pyrazolopyrimidine moiety of AZD5363 is packed between the hydrophobic chains of the top and bottom amino acid residues, forming π-alky interactions with ALA171, MET227, LEU156, VAL164 (top side), and ALA230, MET281(bottom side). The amine pendant of the co-crystal ligand extends towards the solvent-exposed region mounting two H-bond interactions with GLU234 and GLU278 in close proximity to the catalytic site and the entrance channel. Moreover, para-fluorobenzyl moiety remains settled within a shallow hydrophobic cavity in the N-terminal domain forming a halogen interaction with the electropositive carbonyl carbon and the electronegative para-fluoro substituent. The terminal hyroxyethyl extended within the solvent-exposed region establishing a carbon-Hydogen bond with ASN279.
Table 1
Docking score, Glide score, and Glide emodel results of the top 10 docked structures.
Table 2
Ligand interactions of top four compounds.
Based on the lowest docking score the top ten docked ligands are tabulated in Table 1. The chemical structures of top scoring compounds were given in Figure 1. The docking scores are also augmented by the glide score and glide e-model results. Among these selected compounds the top 4 compounds (Irciniastatin B, Irciniastatin A, Pseudoceroxime D, (1'S)-7-chloroaverantin) were subjected to MD-simulation studies in order to validate the findings of the dicking experiment. The ligand Hydrogen bond (H-bond) interactions, MMGBA of the aforementioned four compounds are detailed in the Table 3; Table 2. The compound Irciniastatin B showed better binding within the 4GV1 binding pocket with 7 H-bond interactions compared to the 6 H-bonds formed by the AZD5363 within the same binding site. The lead compound Pseudoceroxime D, with 6 H-bonds showed comparable docking profiles with the AZD5363, co-crystal ligand.
All docked structures occupy the same binding cavity with a few differences in binding interactions. The H-bond interactions in the hinge regions seems imperative for the ligand binding and interestingly enough the H-bond interaction with ALA230 is retained by Irciniastatin B and Pseudoceroxime D, while (1'S)-7-chloroaverantin retains the H-bond interaction with GLU234 in the solvent-exposed region. It is important to note that the lead compounds provided a different binding profile in comparison with the co-crystal ligands as all the H-bonds were formed with residues within the binding pocket. In terms of docking similarity, the ALA230 bond was common with the nascent ligand in 2 lead molecules (Irciniastatin B and Pseudoceroxime D) and the parent interaction of GLU234 was conserved in the docking of (1'S)-7-chloroaverantin, which was also the structure with the minimum number of H-bonds (3 in total). A new H-bond was formed with the GLU228 residue by all the lead compounds and the top three leads also formed an H-bond with LYS179 residue.
This drastic difference in binding profiles from the parent AZD5363 ligand and the lack of commonality between the individual leads too for that matter can be attributed to their widely different chemical structures. Only the isomeric leads Irciniastatin B and Irciniastatin A depicted identical binding profiles with the former having two additional H-bonds than the latter. The shape similarity of a molecule with an existing active molecule or a drug, does not necessarily imply that the new molecule should also possess similar activity potential. However, the shape along with appropriate chemical functionality are key factors for a compound to possess activity against a specific target. Thus, a combination of structural similarity mated with the chemical/steric functionality may allow structurally diverse molecules to exhibit similar biological potential i.e. structurally dissimilar moieties may adopt similar active profiles within the same binding pocket dictated by the steric and chemical interactions mounted within the same binding pocket. This may explain comparable results obtained during both molecular docking and MD simulation experiments as the volume overlap of two molecules is analogous despite the structural diversity.
Table 3
MM-GBSA results of the top four compounds
Compound Name | Docking score (kcal/mol) | MM-GBSA Binding Energy (kcal/mol) |
|---|---|---|
|
|
|
Irciniastatin B | -10.689 | -51.02 |
Irciniastatin A | -8.145 | -37.48 |
Pseudoceroxime D | -7.809 | -37.26 |
(1'S)-7-chloroaverantin | -7.567 | -51.42 |


Table 4
ADME/Tox properties of the top four selected compounds that exhibited a high binding efficiency.




ADMET
The use of computational ADMET screening has the potential to decrease the expenses associated with resource-intensive wet-lab studies, which often result in failure on several occasions (Hage-Melim et al., 2020; Zhang et al., 2020). In the present investigation, it was observed that all of the molecules that were chosen adhered to the optimal threshold of ADMET characteristics. The compounds exhibit alignment within the permissible range of -2.0 to 6.5 for the octanal/water partition coefficient (QPlogPo/w), -6.5 to 0.5 for the aqueous solubility (QPlogS), and -3.0 to 2.1 for the brain/blood partition coefficient (QPlogBB) (Table 4 ) (Gleeson, Bravi, Modi, & Lowe, 2009). The evaluation of drug candidates for their compliance with established criteria of drug-likeness and physiochemical properties is a crucial preliminary screening procedure, with the objective of identifying and eliminating unsuitable candidates (Mohamed, El-Serwy, & El-Serwy, 2021). The Lipinski's rule of five (RO5) is a significant determinant in assessing the drug-likeness of compounds. It assists in the identification of potential molecules from a collection of drug-like substances that should possess favorable characteristics such as robust gastrointestinal absorption, high oral bioavailability, and adequate membrane permeability. These desirable properties are indicated by the following criteria: a logarithm of the partition coefficient (log P) less than or equal to 5, a molecular weight (MW) less than or equal to 500 Da, a number of hydrogen bond donors (HBDs) less than or equal to 5, and a number of hydrogen bond acceptors (HBAs) less than or equal to 10 (Manivannan et al., 2022), and these parameters for the studied compounds were noted to be within the acceptable ranges (Table 4 ).
Dynamic Simulations, Comprehensive Analysis of Structural Flexibility and Stability of top scoring compounds
Top four docking complexes Irciniastatin B-PKBα, Irciniastatin A-PKBα, Pseudoceroxime D-PKBα and (1'S)-7-chloroaverantin- PKBα were post- processed with MD simulations the stability of the studied complexes was elucidated by computing RMSD during simulation. All complexes stable during the simulation period and RMSD remained below 2.5 Å for protein or ligand (Figure 3). Among all four complexes, the least RMSD was found to be displayed by Irciniastatin B in Irciniastatin B-PKBα bonded system (Figure 3A); which confirms that the compound Irciniastatin B is firmly bonded to PKBα. The RMSD curve for protein fluctuated between 1.2 to 2.4 Å, which reflects the conformational changes in protein structure without disrupting the structure. Interestingly, compound Irciniastatin B shows a high level of stability throughout the time duration 100ns, which strongly suggests that the Irciniastatin B maintained its initial conformation throughout the simulation and did not lose any major interaction. The same results are confirmed by the highest value of docking scores. In the case of CP1575/1 bonded system, compound CP1575/1 showed a minor fluctuation during the initial frames of the MD simulation. However, the fluctuation is not too high and the Irciniastatin A-PKBα system acquired equilibrium after 20ns. As shown in Figure 3B, both ligand and protein in the Irciniastatin A-PKBα complex remained stable after 40ns of MD simulation. Among all four complexes (Figure 3) (1'S)-7-chloroaverantin-PKBα complex (Figure 3D) showed the highest fluctuations with the biggest RMSD values of ∼2.1 Å and ∼2.3 Å for protein and ligand, respectively, indicating that (1'S)-7-chloroaverantin is weakly bonded to PKBα (Figure 4B). Pseudoceroxime D has also shown some fluctuation in Pseudoceroxime D- PKBα complex. As shown in Figure 3C, Pseudoceroxime D remained highly stable during initial snapshots (1-65ns), however, a jump of ∼1.1 Å in the RMSD curve was obtained at 67ns and the system immediately acquired equilibrium. Again, these facts are also in accordance with the docking results, which suggest that the compounds Irciniastatin B and Irciniastatin A are more firmly bonded to PKBα than that of Pseudoceroxime D and (1'S)-7-chloroaverantin. Figure 4A-D depicts the RMSF versus the residue number and the RMSF of the PKBα’s residues from the MD trajectory appears same trend which obtained from from the X-ray crystallographic data. This result suggests that the Irciniastatin B exhibited higher conformational changes in the structure of PKBα with M281L.
Molecular docking analysis does not show the relative binding energy or free energy of each ligand or the affinity between the protein and ligand complexes (Pattar et al., 2020). Therefore, an exhaustive analysis of the computed binding affinities of Irciniastatin B, Irciniastatin A, Pseudoceroxime D, and (1'S)-7-chloroaverantin towards PKBα has been performed by employing the MM/PB(GB)SA approach. The obtained binding affinities of ligand-protein complexes were then summarized in Table 2 to compare with the binding affinities of selected molecules bonded to PKBα. The binding free energy values ΔGpred (GB) computed by MM/PB(GB)SA approach reveals that the compounds Irciniastatin B with the highest docking scores also exhibit the highest binding affinity (-29.85 kcal·mol−1) towards PKBα and are more strongly bonded to the studied protein than that of Irciniastatin A, Pseudoceroxime D and (1'S)-7-chloroaverantin. Since Irciniastatin B showed the most significant difference in ΔGpred values for PKBα in contrast to other compounds; it reflects that the compound Irciniastatin B is the most potent PKBα inhibitor among all four inhibitors. Similarly, in the PKBα -bonded system, Irciniastatin A, and Pseudoceroxime D demonstrate comparable binding affinities to each other (ΔGpre –37.48 and -37.26 kcal∙mol-1, respectively) for PKBα. Last but not the least, compound (1'S)-7-chloroaverantin has also demonstrated appreciable affinity towards PKBα (ΔGpre –37.48 kcal∙mol-1). Taking together, MMGBSA results are in strong correlation with the findings of docking analysis and suggest that the Irciniastatin B could be the most potent PKBα inhibitor among all four selected compounds. Although, compound(1'S)-7-chloroaverantin shares the least binding affinity in terms of docking scores, high MMGBSA binding affinity presents (1'S)-7-chloroaverantin as a potential PKBα inhibitor.
To pinpoint the essential amino acids that effectively maintained contact with ligands within the PKBα complex, we generated a timeline representation illustrating various interactions such as hydrogen bonds, hydrophobic interactions, ionic interactions, and water bridges, as shown in Figure 5. Notably, during both docking and MMGBSA analysis, Irciniastatin B exhibited the highest binding affinity for PKBα. These findings were further substantiated by the protein-ligand contact analysis chart, as illustrated in Figure 4A, where Irciniastatin B consistently established and sustained approximately twelve interactions with adjacent residues. Within the Irciniastatin B- PKBα complex system, amino acids T160, M227, A230, Y272, D274, K276, E278, N279, and D292 emerge as the pivotal residues, consistently maintaining contact with the ligand for more than 80% of the observed duration. The top panel shows the total number of specific contacts the protein makes with the ligand over the course of the trajectory. Conversely, Irciniastatin A, Pseudoceroxime D, and (1'S)-7-chloroaverantin in their corresponding complexes were able to establish and maintain at least eight contacts with the closely available residues (Figure 5B-D). It's intriguing to observe that certain amino acid residues, namely E228, A230, E234, E278, and D292, exhibit a consistent interaction pattern across all studied ligands Irciniastatin B, Irciniastatin A, Pseudoceroxime D, and (1'S)-7-chloroaverantin when bound to PKBΑ. These residues seem to play a key role in forming stable contacts with the ligands. Moreover, the residue D292, which is conserved across these systems, stands out as it forms more than one specific interaction with the ligands in all of the analyzed cases. This enhanced level of interaction is represented graphically by a darker shade of orange on the colour scale located to the right of the plot, highlighting the significance of this particular residue in the binding process. In summary, the results from our current analysis align well with the docking outcomes, confirming that all four chosen ligands exhibit significant binding affinities for PKBα. Notably, Irciniastatin B stands out as it demonstrates superior binding affinity compared to the other ligands studied, primarily due to its ability to establish additional interactions with neighbouring residues.
Conclusion
This study undertook a comprehensive investigation of natural substances as possible inhibitors of Protein Kinase B-alpha (PKBα), a critical target in the field of cancer treatment. By employing virtual screening and employing structure-based predictions, we have identified Irciniastatin B as a very promising candidate for therapeutic development. This compound has exceptional binding affinity and establishes a strong network of interactions with PKBα. The results of our study are substantiated by employing a comprehensive approach that includes molecular docking, MM/GBSA binding energy calculations, molecular dynamics simulations, and ADMET evaluations. The confirmation of the stability of PKBα-ligand complexes was achieved by conducting thorough molecular dynamics simulations, which emphasized the persistent connections between the ligands and crucial residues of the protein. Moreover, there is a good correlation between the anticipated binding affinities and the docking scores, which serves to enhance the credibility and dependability of our findings. In conclusion, our study showcases the potential of naturally occurring chemicals, specifically Irciniastatin B, as very promising contenders for the advancement of PKBα inhibitors in the realm of cancer therapy. The aforementioned discoveries not only contribute to the advancement of our comprehension of the molecular interactions implicated, but also present novel opportunities for the exploration of therapeutic agents in the ongoing battle against cancer. The ongoing exploration of plant-derived natural chemicals as inhibitors of cancer cell proliferation is a highly competitive and productive field of research, holding promise for the development of more efficacious cancer treatments in the coming years.
Author contributions
Conceptualization, K.R.R.R; writing-original manuscript, KRRR, HS; Methodology, data curation, and formal analysis, KRRR, BB; Project administration and Supervision, K.R.R.R; Review and editing, KRRR, BB, HS.; Interpretation, and review/revision, B.B and K.R.R.R. All authors have read and agreed to the published version of the manuscript.